用于CRISPR的gRNA的设计
前排:我这都是抄的,你要转发引用什么的千万别说是从我这里看到的,去引用原文链接。
How to Design Your gRNA for CRISPR Genome Editing (addgene.org)
Originally published May 3, 2017 and last updated Sep 24, 2020
This post was contributed by guest blogger, Addgene Advisory Board member, and Institute Scientist at the Broad Institute, John Doench.
CRISPR technology has made it easier than ever both to engineer specific DNA edits and to perform functional screens to identify genes involved in a phenotype of interest. This blog post will discuss differences between these approaches, and provide updates on how best to design gRNAs. You can also find validated gRNAs for your next experiment in Addgene’s Validated gRNA Sequence Datatable. A more extended discussion of these subjects can be found in two recent review articles (Doench et al., 2017, and Hanna et al., 2020) and references therein.
mportant considerations before you start an experiment with CRISPR
The hammer, the jigsaw, and the wrench are all great tools, but which one you use, of course, depends on what you are trying to do – there’s no “best” tool among them. While this seems obvious, it is important to remember that the same is true when designing gRNAs for using CRISPR technology – the “best” gRNA depends an awful lot on what you are trying to do: gene knockout, a specific base edit, or modulation of gene expression.
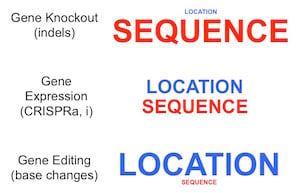 Location and sequence are important considerations for designing your gRNAs. For indels, it’s not so important what location in the gene you target, but it is important that your gRNA sequence is designed to be highly active and reduce off targets. For CRISPRa and CRISPRi, these considerations are of roughly equal importance (target should be near the TSS but you can worry less about sequence optimality because you generally have fewer sequences to choose from). Finally, for HDR, location is much more important because you have to target within ~30 nt of your proposed edit, which means there are so few gRNAs to choose from that sequence preferences must largely be ignored.
Location and sequence are important considerations for designing your gRNAs. For indels, it’s not so important what location in the gene you target, but it is important that your gRNA sequence is designed to be highly active and reduce off targets. For CRISPRa and CRISPRi, these considerations are of roughly equal importance (target should be near the TSS but you can worry less about sequence optimality because you generally have fewer sequences to choose from). Finally, for HDR, location is much more important because you have to target within ~30 nt of your proposed edit, which means there are so few gRNAs to choose from that sequence preferences must largely be ignored.
The hammer: Gene knockout by NHEJ
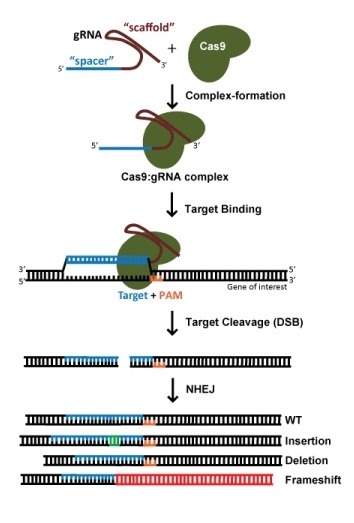 Gene knockout with CRISPR technology is usually accomplished by Cas9-mediated dsDNA breaks: following a cut, the error-prone nature of non-homologous end joining (NHEJ) often leads to the generation of indels and thus frameshifts that disrupt the protein-coding capacity of a locus. When using S. pyogenes Cas9 (SpCas9), potential target sites are both [5’-20nt-NGG] and [5’-CCN-20nt], as it is equally efficacious to target the coding or non-coding strand of DNA. As a rule of thumb, we avoid target sites that code for amino acids near the N’ terminus of the protein, in order to mitigate the ability of the cell to use an alternative ATG downstream of the annotated start codon. Likewise, we avoid target sites that code for amino acids close to the C’ terminus of the protein, to maximize the chances of creating a non-functional allele. For a 1 kilobase gene, since potential target sites occur ~1 in every 8 nucleotides, restricting gRNAs to 5 – 65% of the protein coding region will still result in many dozens of gRNAs to choose from. With so many possibilities, picking a gRNA with an optimized sequence is of primary importance (more on this below).
Gene knockout with CRISPR technology is usually accomplished by Cas9-mediated dsDNA breaks: following a cut, the error-prone nature of non-homologous end joining (NHEJ) often leads to the generation of indels and thus frameshifts that disrupt the protein-coding capacity of a locus. When using S. pyogenes Cas9 (SpCas9), potential target sites are both [5’-20nt-NGG] and [5’-CCN-20nt], as it is equally efficacious to target the coding or non-coding strand of DNA. As a rule of thumb, we avoid target sites that code for amino acids near the N’ terminus of the protein, in order to mitigate the ability of the cell to use an alternative ATG downstream of the annotated start codon. Likewise, we avoid target sites that code for amino acids close to the C’ terminus of the protein, to maximize the chances of creating a non-functional allele. For a 1 kilobase gene, since potential target sites occur ~1 in every 8 nucleotides, restricting gRNAs to 5 – 65% of the protein coding region will still result in many dozens of gRNAs to choose from. With so many possibilities, picking a gRNA with an optimized sequence is of primary importance (more on this below).
The jigsaw: Editing by HDR, base editing, and prime editing
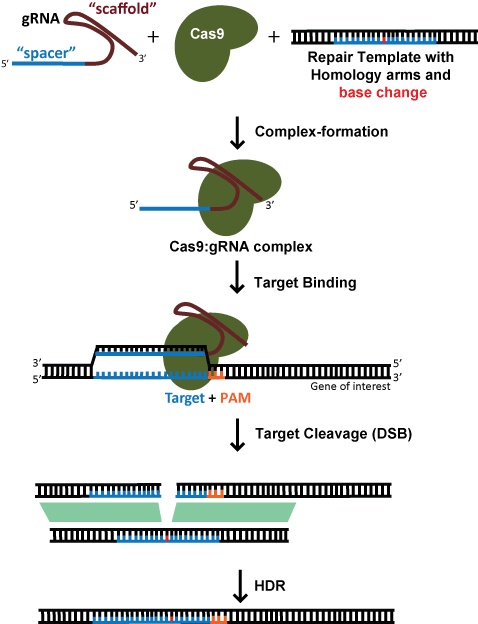
For a specific edit, such as the insertion of a fluorescent tag or the introduction of a specific mutation, one generally relies on homology directed repair (HDR) to incorporate new information into DNA. This also requires an exogenous DNA template. HDR, however, is a very low-efficiency process, and usually involves the need for single cell cloning and subsequent screening for successful edits. This is a very time consuming process and should not be undertaken lightly! Indeed, truly achieving the gold standard requires not one but two rounds of single cell cloning – as a control, one should revert the edit back to the original in order to prove that the phenotype was really due to the intended edit rather than some passenger variant that came along with the single cell clone (although this is rarely done).
When targeting a dsDNA break for HDR, the choice of target site is far more constrained by the desired location of edit; efficiency decreases dramatically when the cut site is >30nt from the proximal ends of the repair template (Yang et al., 2013). This means that, for gene editing, there are usually few potential gRNAs. While SpCas9, with a PAM preference of NGG, is still the most widely-used Cas enzyme, the development of SaCas9, NmeCas9, Cas12a enzymes, and engineered variants thereof offers additional PAM options that can greatly expand gRNA options.
Two newer technologies offer an alternative to HDR for introducing edits. The same locational constraints are even more exquisite for the so-called base editor Cas9, which makes DNA changes in the absence of dsDNA breaks (Rees et al., 2018). For C>T and A>G base editors, the intended edit must be in a 5 - 10 nt window relative to the PAM, and bystander edits are possible if there is another target C or A in the window. Another technology, prime editing (reviewed in Anzalone et al., 2020). is not limited to single nucleotide transitions but still requires a nearby PAM, although these are still early days for this technology, and the user may need to optimize numerous parameters to generate the desired edit.
The wrench: Gene activation and inhibition by CRISPRa and CRISPRi
Finally, for modulating gene expression at the level of transcription – CRISPRa (activation) and CRISPRi (inhibition) technologies – a nuclease-dead Cas9 (dCas9) is directed near the promoter of a target gene. Here, the target window is not quite as broad as for knockout via CRISPR cutting. For CRISPRa, it is most-efficacious to target a ~100nt window upstream of the transcription start site (TSS), while for CRISPRi, a ~100nt window downstream of the TSS gives the most activity. Thus, a given gene will only have a dozen or so gRNAs to choose from in the optimal location. It is also important to have good information on the exact location of the TSS. Different databases annotate the TSS in different ways, and it has been shown that the FANTOM database, which relies on CAGE-seq to directly capture the mRNA cap, provides the most accurate mapping (Radzisheuskaya et al., 2016). In this case, location and sequence are of approximately equal importance in design – an optimized sequence will do little if it is in the wrong place, but because the target window is more-narrow, there are fewer gRNA to choose from, and thus an optimal sequence may not be available.
Predicting gRNA efficacy
We and others have examined the ability to use sequence-based and other features to nominate gRNAs that are likely to be active, not only for SpCas9 but also for some other Cas enzymes. It seems to be the case that there is no universal scoring system for selecting a gRNA, as the method of producing the guide (synthetic, in vitro transcription, or lentiviral delivery) can affect the accuracy of a predictive score, as well as dynamic aspects of the target (e.g. accessibility due to chromatin status). No computational prediction is ever perfect, but this can decrease the number of guides one needs to test in the lab.
Importantly, for any modification of interest, it would be unwise to make conclusions on the basis of the activity of a single gRNA, and thus diversity of gRNAs across a gene should be examined whenever possible when using knockout or transcriptional modulation approaches.
Avoiding off-target effects

The off-target activity of gRNAs is important to consider. While the basic landscape of mismatches that can nevertheless still lead to activity has been established, and can be used to identify sites that are likely to give rise to an off-target effect, there’s not enough data to fully predict which sites will and will not show appreciable levels of modification. Whole-genome sequencing of cells modified by CRISPR indicates that the consequences of off-target activity, at least for the experimental conditions used, led to no detectable mutations (Veres et al., 2014). When working with single-cell clones, the authors note that “clonal heterogeneity may represent a more serious obstacle to the generation of truly isogenic cell lines than nuclease-mediated off-target effects.” Further, large-scale datasets of hundreds of genetic screens using genome-wide libraries have shown high concordance between different sequences targeting the same gene, suggesting that off-target effects did not overwhelm true signal in these assays (Dempster et al., 2019). Again, the experimental strategy is clear: for any gene of interest, one should require that multiple gRNAs of different sequences give rise to the same phenotype in order to conclude that the phenotype is due to an on-target effect.
Conclusions
Selection of gRNAs for an experiment needs to balance maximizing on-target activity while minimizing off-target activity, which sounds obvious but can often require difficult decisions. For example, would it be better to use a less-active gRNA that targets a truly unique site in the genome, or a more-active gRNA with one additional target site in a region of the genome with no known function? For the creation of stable cell models that are to be used for long-term study, the former may be the better choice. For a genome-wide library to conduct genetic screens, however, a library composed of the latter would likely be more effective, so long as care is taken in the interpretation of results by requiring multiple sequences targeting a gene to score in order to call that gene a hit.
This is an exciting time for functional genomics, with an ever-expanding list of tools to probe gene function. The best tools are only as good as the person using them, and the proper use of CRISPR technology will always depend on careful experimental design, execution, and analysis.

Many thanks to our guest blogger John Doench!
 John Doench is the Director of R&D in the Genetic Perturbation Platform at the Broad Institute and has worked with many Addgenies to help improve the understanding, curation, and explanation of our CRISPR resources. He really likes small RNAs.
John Doench is the Director of R&D in the Genetic Perturbation Platform at the Broad Institute and has worked with many Addgenies to help improve the understanding, curation, and explanation of our CRISPR resources. He really likes small RNAs.
 wechat
wechat
